MedFriendly®
















Stevens-Johnson Syndrome
Stevens-Johnson syndrome is a rare but serious
condition in which the skin and at least two surfaces of
the mucous membranes (or the mucous membranes
only) are damaged by a severe reaction to infection or
medication. A mucous membrane is one of four major
types of thin sheets of tissue that line or covers various
parts of the body.
WHAT ARE THE SIGNS AND SYMPTOMS OF
STEVENS-JOHNSON SYNDROME?
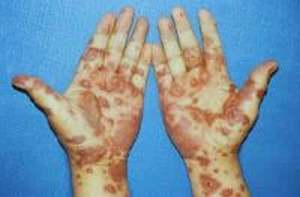
Skin damage from Stevens-
Johnson syndrome.
FEATURED BOOK: Lippincott Pocket Drug Guide for Nurses
chills, sore throat, fever, cough with a thick pus-like sputum [spit], fatigue, malaise [feeling
unwell], breathing difficulties), arthrlagias (joint pains), and burning eyes. The initial signs
and symptoms are usually mistaken for infection and treated with antibiotics, which can
actually make the condition worse. The initial signs and symptoms are followed 1 to 14
days later by signs of a painful, burning, red or purple blistery rash on flat, round,
thickened areas of skin about an inch across (e.g., like hives) in the face and upper trunk
that spreads throughout the body (arms, legs, soles of the feet) within hours to days.
Sometimes only one area of skin may be affected, usually the trunk. The rash can begin
as flat discolorations that develop into: a) small raised areas of skin with no visible fluid,
b) small sacs that contain fluid, c) a bubble-like area filled with air or fluid, d) a target-like
appearance, e) red areas with a smooth surface, or f) red areas of skin that combine
together. There are usually two zones of color.
"Where Medical Information is Easy to Understand"™
Between 10 to 90% of the skin can be involved in Stevens-Johnson
syndrome. This will appear quite gruesome because the top layer of
skin (the epidermis) will eventually die and shed off. This can
happen when skin lesions rupture and these areas usually become
infected and lead to scarring. Some cases can be milder than
others. The face and tongue can swell. The mucous membranes of
the mouth (including the lips), nose, and eyes are particularly
affected with a rash and/or blisters. The mouth and lips are almost
always affected. The mouth involvement is usually very painful and
decreases the ability to eat and drink. Tissue death (ulcers) occurs
extensively in these areas and the skin can peel away in sheets in
a basket weave-like pattern.
There is minimal associated inflammation and swelling (edema) and the area usually appears red. The
genital area and anal area can be affected as well. The hair and nails can fall out in some cases. The
mucous membranes of the lower respiratory tract can be affected.
Tissue death can also occur in the gastrointenstinal and respiratory tissues. The skin on the outer arms,
legs, hands, feet, knees, elbows, etc., are affected more than the inner skin areas of these body parts
(e.g., palms, soles).
A purulent (pus producing) conjunctivitis can occur such as in 30% of children who develop Stevens-
Johnson syndrome. Conjunctivitis is inflammation of the conjunctiva. The conjunctiva is a layer that
covers and protects the inside of the eyelids and the front part of the sclera (the white part of the eyes).
Conjunctivitis can be either papillary or follicular. If it is papillary, the conjunctiva will have a cobblestone
appearance of flattened nodules and a core of blood vessels. If it is follicular, the nodules in the
conjunctiva will appear dome-shaped without a prominent central blood vessel. The conjunctiva can shrink
in Stevens-Johnson syndrome and areas below the top layer can become thickened and hardened.
Foreshortening of the fornix of the conjunctiva can occur. The fornix of the conjunctiva connects the part
of the conjunctiva that lines the eyelids and the part that covers the eyeballs. Sometimes, these two parts
of the conjunctiva can partly or completely stick to each other in Stevens-Johsnon syndrome, which is
known as symblepharon. There can also be ankyloblepharon, which is when edges of the eyelid stick to
each other.
In some cases, conjunctivitis in Stevens-Johnson syndrome can lead to open sores in the eyes and
formation of scar tissue inside the eyelids, leading to vascularization (excessive growth of blood vessels)
of the cornea. The cornea is the clear covering at the front of the eyeball. Tightly packed skin cells
covering the cornea (known as epithelial cells) can be damaged. Ulcers of the connective tissue of the
cornea can also occur. The cornea can also become perforated and/or it can become cloudy, which
impairs vision. Keratitis (inflammation of the cornea), iritis (inflammation of the iris), uveitis (inflammation
of the uvea), and sometimes blindness can occur. The iris is the colored part of the eye. The uvea is the
middle layer of the eye. Limbitis can also occur, which is inflammation of the cornea of the limbus. The
corneal limbus is the border of the cornea and the sclera (the white part of the eye).
Other eye problems can occur in Stevens-Johnson syndrome such as blepharitis (inflammation of the
eyelids), trichiasis (abnormally positioned eyelids that grow backwards towards the eyes), and distichiasis
(abnormal growth of eyelashes from the rim of the eyelids at the opening of the meibomian glands). The
meibomian glands produce an oily substance in the eyes and can be dysfunctional in Stevens-Johnson
syndrome.
WHAT CAUSES STEVENS-JOHNSON SYNDROME?
In most cases, Stevens-Johnson syndrome is caused by a hypersensitive allergic reaction to medication,
infection, illness, and rarely by cancer. This is caused by a fault in the bodys immune (defense) system.
When medications are the cause, signs usually begin within a week and the specific medication triggering
the reaction is usually an antibiotic (e.g., tetracycline, amoxicillin, ampicillin, levofloxacin, azithromycin,
cefixime, ciprofloxacin, trimethoprim, sulfonamides, penicillin). Non-steroidal anti-inflammatory drugs
(NSAIDs, e.g., ibuprofen, valdecoxib, diclofenac, piroxicam, meloxicam, tenoxicam), which are
medications used to treat pain and inflammation are rare causes of Stevens-Johnson syndrome but the
risk is higher in older adults and in those beginning treatment.
Another group of medications that can cause Stevens-Johnson syndrome is those used to treat gout
(such as allopurinol) which is a type of arthritis. In fact, allupurinol is the most common cause of Stevens-
Johnson syndrome in Southeastern countries such as Singapore, Malaysia, Hong Kong, and Taiwan.
Anticonvulsants (medications used to treat seizures; e.g., phenytoin, carbamazepine, oxcarbazepine,
ethosuxamide, zonisamide, phenobarbital, valproate, lamotrigine) are also known to cause Stevens-
Johnson syndrome. Combining the latter two medications is known to increase the risk. When anti-
convulsants cause Stevens-Johnson syndrome, it is usually caused in the first 60 days.
Other medications/drugs that can cause Stevens-Johnson syndrome include medications to treat viral
infections (e.g., etravirine, oseltamivir, nevirapine, telaprevir, indinavir), medication used to treat acne
(e.g., isotretinoin), anti-depressant medication (e.g., bupropion, mirtazapine, sertraline), anti-fungus
medications (e.g., fluconazole, nystatin), stimulant medications (e.g., modafinil), pain killers (e.g.,
Tramadol), cocaine, some over the counter medications (e.g., acetaminophen, NSAIDs), anti-parasite
medications (e.g., pyrimethamine), cough and cold medications, medications used to treat acid reflux
(e.g., pantoprazole), barbiturates, medications used to treat diabetes mellitus (e.g., sitagliptin), and tumor
necrosis factor(TNF)-alpha antagonists (eg, infliximab, etanercept, adalimumab). Barbiturates are relaxing
drugs that act by depressing breathing rate, blood pressure, and brain functioning. Some barbiturates are
also used to treat seizures. Diabetes mellitus is a medical condition in which the body is not able to
effectively use a natural chemical called insulin. Insulin's main job is to quickly absorb glucose (a type of
sugar) from the blood into cells for their energy needs and into the fat and liver (a large organ that
performs many chemical tasks) cells for storage. TNF-alpha antagonists are a class of medications that
reduces inflammation.
Viral and bacterial infections can sometimes (but not often) cause Stevens-Johnson syndrome including
the flu, mycoplasmae pneumoniae (a small type of bacteria that cause pneumonia), yersinia (a type of
bacteria), hepatitis (an infection of the liver that causes liver inflammation), herpes (a type of virus such
as herpes simplex or herpes zoster), and HIV (human immunodeficiency virus)/AIDS (acquired immune
deficiency syndrome). HIV is a virus that attacks the body's immune system, leading to infections and
harmful tumors. AIDS is a decrease in the effectiveness of the body's immune (defense) system that is
due to HIV infection.
Other infectious causes of Stevens-Johnson syndrome includes Epstein-Barr virus (one of the most
common viruses in humans), typhoid, diphtheria , mumps, cat-scratch fever, coxsackie virus, Group A
beta-hemolytic streptococci, brucellosis, lymphogranuloma venereum, mycobacteria, rickets, and
tularemia.
A brief description of these conditions follows (skip to the next paragraph if not interested). Typhoid is a
bacterial disease that typically affects the intestines. Diphtheria is a suddenly occurring, contagious and
infectious disease due to the bacteria, Corynebacterium diphtheriae, and its very strong toxin. Mumps is
a type of virus that occurs suddenly, mainly in children, and usually causes the appearance of swelling in
the face. Cat-scratch fever is a usually benign infectious disease caused by the bacteria Bartonella
hensellae, usually in children a week or two after a cat scratch or cat bite. The coxsackie virus is a
common human virus. It is a type of enterovirus, which is a very common group of viruses affecting
humans. Enteroviruses also put people at risk for developing Stevens-Johnson syndrome. Group A beta-
hemolytic streptococci is a form of bacterial infection that causes many types of invasive and non-
invasive infections. Brucellosis is a highly contagious disease spread between non-human animals to
animals or vice versa. Lymphogranuloma venereum is a type of sexually transmitted disease.
Mycobacteria is a type of bacteria that grows in a mold-like fashion when cultured. Rickets is a condition
in which abnormal bone formation is caused by a deficiency of calcium and vitamin D. Tularemia is
serious infection caused by the bacterium, Francisella tularensis.
Fungal infections that can cause Stevens-Johnson syndrome include coccidioidomycosis,
dermatophytosis, and histoplasmosis. Coccidioidomycosis is a fungal infection caused by the fungus,
Coccidioides, which is found in the soil of dry, low rainfall areas. Dermatophytosis is a type of fungus
infection that affects humans, pets, and domesticated animals. Histoplasmosis is a disease caused by the
fungus, Histoplasma capsulatum. Other infectious causes of Stevens-Johnson syndrome include malaria
and trichomoniasis. Malaria is a serious disease caused by parasites that is spread by mosquitoes.
Trichomoniasis is a type of sexually transmitted disease caused by a parasite. A parasite is an organism
that lives in or on another organism to obtain nourishment.
Systemic lupus erythematosus (SLE) can also cause Stevens-Johnson syndrome. SLE is a long-term
disease in which the connective tissues throughout the body are inflamed because the body's defense
system attacks these tissues as if they were foreign substances. People with SLE or HIV are more likely
to develop Stevens-Johnson syndrome caused by medications.
Stevens-Johnson syndrome can also be caused by radiation therapy or ultraviolet light. Radiation for
brain tumors when combined with anticonvulsant treatment also puts one at greater risk for Stevens-
Johnson syndrome. Sometimes, the cause of Stevens-Johnson syndrome cannot be identified.
WHAT ARE RISK FACTORS FOR STEVENS-JOHNSON SYNDROME?
Stevens-Johnson is unpredictable and rare. This makes it difficult to identify risk factors for the condition
although using medications from the classes described above and having the types of infections or
conditions described above leads to an increased risk. People with weakened immune systems are more
likely to develop Stevens-Johnson syndrome. People who have a liver that cannot completely detoxify
reactive chemical byproducts of medications are also at increased risk for Stevens-Johnson syndrome.
Using higher doses of allopurinol and lamotrigine, along with rapidly introducing these medications to the
body, can increase the risk of developing Stevens-Johnson syndrome. More than half of people who
develop Stevens-Johnson syndrome have had a recent upper respiratory infection.
Research has shown that in some East Asian populations (Thai and Hans Chinese) possessing a certain
type of gene puts one at greater risk for developing Stevens-Johnson syndrome. The gene involves
various forms of HLA-B (major histocompatibility complex, class I, B). This gene provides instructions for
making a protein that plays a very important role in the immune system. In Eastern Asian populations, the
HLA-B*1502 (HLA-B75) gene is associated with hypersensitivity reactions to the anticonvulstants
carbamazepine and phenytoin. In Europe, the HLA-B5801 gene is associated with hypersensitivity to
allopurinol (61% of people with hypersensitivity reactions to this medication carried the gene).
Other versions of the HLA gene that make people more prone to developing Stevens-Johnson syndrome,
includes: HLA-B*5801, HLA-B*44 (especially Caucasians), HLA-A29, HLA-B12, HLA-DR7, HLA-A2, HLA-
A*0206, and HLA-DQB1*0601.Having the HLA-A29, HLA-B12, and HLA-DR7 genes are often associated
with Stevens-Johnson syndrome caused by a type of antibiotics known as sulfonamides. Having the HLA-
A2 and HLA-B12 genes are often associated with Stevens-Johnson syndrome caused by NSAIDs. Having
the HLA-A*0206 and HLA-DQB1*0601 is often associated with Stevens-Johnson syndrome that includes
eye disease.
While having one of the HLA genes described above is associated with Stevens-Johnson syndrome, the
presence of the gene(s) is not necessary or sufficient to develop Stevens-Johnson syndrome. It is not
known if having one of these genes predisposes one to Stevens-Johnson syndrome.
HOW IS STEVENS-JOHNSON SYNDROME DIAGNOSED?
The signs from the physical examination are the easiest way to identify Stevens-Johnson syndrome,
especially when combined with a good medical history. The diagnosis can be confirmed by a taking a skin
biopsy (removing a piece of skin tissue) and examining it under the microscope. When examined under
the microscope, mild inflammatory cells will be present, mostly superficial and near the blood vessels.
There will usually also be tissue death of the outer skin layer. The tissue death is primarily caused by the
death of keratinocytes, which are the main type of cell that makes up the outer layer of the skin.
The connection between the outer layer of skin and the layer of skin can have changes in it ranging from
blister formation to alteration of vacuoles (enclosed compartments in cells that contain water and other
molecules). Other findings can be the presence of T-cells (e.g., CD4 positive and CD8 positive) in the
outer layer of skin and the connection area between the outer layer of skin and the layer below it. There
are mostly CD8 positive T-cells in this connection area. A T-cell is a type of white blood cell that directs
the body s immune (defense) system to defend against bacteria and other harmful cells. See the technical
mechanisms section below for why they are present in Stevens-Johnson syndrome.
When the conjunctivae of the eyes are examined in patients who have disease of the eyes, lymphocytes
(types of white blood cells) can infiltrate the area and can also be present around vessel walls. The main
type of lymphocyte that infiltrates these areas is known as the helper T-cell. Plasma cells (other types of
white blood cells) may also be found below the outer layer of skin. Many HLA-DR-positive cells (see prior
section for explanation of these cells) may be present in parts of the conjunctiva and cornea such as the
vessel walls.
HOW IS STEVENS-JOHNSON SYNDROME TREATED?
Stevens-Johnson syndrome is a medical emergency and usually requires hospitalization, usually in a burn
unit or an intensive care unit. Even though the patient is not burned, burn units have expertise in treating
patients with the open skin wounds that occur in Stevens-Johnsons syndrome. Presence of the signs
above should trigger the patient to seek emergency medical care. Once the cause is identified (e.g., a
medication), efforts are undertaken to stop it (e.g., discontinue the medication), control the symptoms, and
decrease complications. If medications are suspected as the cause, the doctor may suggest stopping all
medications to control the signs of Stevens-Johnson syndrome.
Although there are no standard care treatments for Stevens-Johnson syndrome, supportive treatment
during hospitalization includes wounds care, airway maintenance, fluid replacement and nutrition, use of
intravenous pain medications, eye care, and keeping the patient in a warm environment. Due to the risk of
eye problems, an ophthlamologist (type of eye doctor) is immediately consulted to evaluate the patient.
For wound care, cool wet compresses help soothe blisters while they heal. Skin doctors and surgeons
tend to disagree on whether dead skin should be gently removed and covered with a bandage containing
pain relieving medication (if needed). However, most seem to agree that this is needed. The bandage
often contains saline (a salt water solution) or Burows solution (an antibacterial liquid that can also shrink
body tissue). Because the skin contains fluid, significant fluid loss can occur when the skin comes off.
This is why replacing fluids is important. If the patient is unable to drink, he/she may receive fluids through
a tube that is placed in the nose and advanced into the stomach (nasogastric tube) or directly into the
stomach (gastric tube).
Because it is painful to take in food by mouth, a mouth wash can be used that has a pain killing
component. Topical pain medications can also be applied. Prevention of tetanus is also important as it
can enter open areas of skin. Tetanus is a potentially deadly infection of the brain and spinal cord
produced by poison (tetanospasmin) produced by the bacteria, Clostridium tetani.
Eye care involves lubricating the surface of the eye, treatment with antibiotics, topical corticosteroids
(types of medications that reduce inflammation), and separating the two layers of the conjunctiva if they
stick together. In severe cases, surgical removal of hardened areas from the back lid margins is
performed. Grafting of mucous membranes and/or amniotic membranes may be needed to replace areas
of damaged tissue. The amniotic membrane comes from the placenta and can be used to replace
damaged eye tissue. The placenta is an organ in the uterus (a hollow organ in which a baby develops)
that links the blood supply of the mother to the developing baby and by which the baby can release
wastes. Cells from the corneal limbus may also be transplanted. The corneal limbus is the border of the
cornea and the sclera (the white part of the eye).
Although medication can cause Stevens-Johnson syndrome, some medications are used to treat it. This
includes pain medication, antibiotics, antihistamines (medication that helps decrease itching), and topical
steroids (medications that help reduce skin inflammation). Some adults may receive intravenous
corticosteroids, which can decrease recovery time if started a day or two after signs of Stevens-Johnson
syndrome first appear. However, the use of corticosteroid treatment for Stevens-Johnson syndrome is
controversial because there are no randomized controlled trials supporting their use (although this is very
difficult to do with a rare condition) because some studies have shown more complications and longer
hospital stays when corticosteroids are used, and because the condition can be successfully managed
without corticosteroids. For children, use of intravenous corticosteroids can increase the risk of
complications. Intravenous immunoglobulin (IVIG) is a medication that contains antibodies that can help
the bodys immune (defense) system stop the process of Stevens-Johnson syndrome. For people infected
with mycoplasma (a type of bacteria that lacks a cell wall), this can be treated with the oral antibiotics
doxycycline or macrolide. Some people have been treated with medications used to treat immune
disorders such as cyclophosphamide or cyclosporine but neither have been very successful.
Rarely, if a large area of skin is affected, a skin graft may be performed in which skin is removed from one
part of the body and attached to the injured area. Sometimes, a synthetic skin substitute may be used for
the skin graft. Often, however, the skin is left to regrow on its own.
WHO USUALLY GETS STEVENS-JOHNSON SYNDROME?
Stevens-Johnson syndrome is most common in older people, although it can happen at any age. Older
people are more prone to develop the condition because they are more likely to use the medications that
cause it. Some have reported that twice as many women are likely to develop the condition than men.
However, some studies report that the difference is not always quite that high (e.g., 62% female) whereas
others report fewer women than men with the condition (e.g., 33% female). Cases of Stevens-Johnson
syndrome tend to happen in early spring and winter. It is more common in Caucasians. Children as young
as 3-months of age can develop Stevens-Johnson syndrome. The average age of someone with Stevens-
Johnson differs from study to study, from early adulthood (e.g., age 25) to middle adulthood (e.g., age 47).
HOW MANY PEOPLE DEVELOP STEVENS-JOHNSON SYNDROME PER YEAR?
Stevens-Johnson syndrome is rare, only affecting between 2.6 to 7.1 individuals per million people a year.
In the United States, about 300 new cases a year are diagnosed. One exception to the yearly incidence
rate appears to be Germany, with a reported number of 1.1 cases per million people a year.
CAN NON-HUMAN ANIMALS DEVELOP STEVENS-JOHNSON SYNDROME?
Yes, some non-human animals such as dogs, cats, and monkeys can develop Stevens-Johnson
syndrome.
CAN STEVENS-JOHNSON SYNDROME BE PREVENTED?
If a medication has been identified as causing Stevens-Johnson syndrome, it should be avoided
permanently in the future. Starting does of allopurinol and lamotrigine at lower levels and increasing the
dose slowly will lessen the risk of developing Stevens-Johnson syndrome. The U.S. Food and Drug
Administration and Health Canada advises screening patients of southeastern Asian ethnicity for the HLA-
B*1502 gene before beginning treatment with carbamazepine.
In August 2013, based on a review of medical literature and cases of adverse reactions, the U.S Food
and Drug Administration announced that any person developing a rash, blister, or other skin reaction while
using acetaminophen should stop using the drug and receive immediate medical care.
HOW IS STEVENS-JOHNSON SYNDROME CLASSIFIED?
Since 1983, Stevens-Johnson syndrome has traditionally been considered to be a variant of erythema
multiforme major, which is an acute (sudden), self-limited, and sometimes recurring skin condition that is a
hypersensitivity reaction associated with certain medications, infections, and other triggers. Erythema
multiforme is usually caused by an infection, usually from the herpes simplex virus. Unlike Stevens-
Johnson syndrome, erythema multiforme is usually benign.
In the 1990s, some scientists proposed that Stevens Johnson syndrome is separate from erythema
multiforme major, with the latter being restricted to cases where the skin lesions have a target appearance
(pink red ring around a lighter center) or raised and swollen, small, enlarged abnormal areas of tissue (with
or without mucosal membrane involvement). Unlike the classic target appearing lesions in erythmea
multiforme major, it has been proposed that Stevens-Johnson syndrome diagnoses should be restricted to
those with damage to the mucous membranes, widespread small blisters, or red bleeding areas of skin.
Thus, it is the pattern of skin lesions that would differentiate the two disorders according to this
classification system.
Some scientists consider Stevens-Johnson syndrome a less severe form of toxic epidermal necrolysis
(TEN), which is a rare and sometimes life threatening disorder of the skin caused by a fault in the immune
system. Some have suggested that the term Stevens-Johnson syndrome be reserved for cases in which
less than 10% of the total body surface is involved, that TEN be used for cases in which more than 30%
of the total body surface is involved, and that overlapping Stevens-Johnson syndrome/TEN be used for
cases in which between 10 and 30% of the total body surface is involved. Stevens-Johnson syndrome
and TEN were first recognized in 1922. Some have supported this unification by saying that erythema
multiforme is mostly caused by the herpes virus and that Stevens-Johnson syndrome and TEN are usually
caused by medications (with more potentially dangerous consequences). Although some have claimed
that Stevens-Johnson syndrome and erythema multiforme (but not TEN) are often caused by the herpes
virus, other reports show that herpes does not often cause Stevens-Johnson syndrome.
WHAT IS THE PROGNOSIS FOR PEOPLE WITH STEVENS-JOHNSON SYNDROME?
It can take weeks to months to recover from Stevens-Johnson syndrome. However, the outer layer of skin
can begin to regrow in days after the cause is eliminated and the skin reaction is stopped. The skin
usually heals within one to three weeks (unless secondary infection occurs). Some areas of skin such as
mucous membranes may remain eroded and crusted for two weeks or more. If the herpes virus caused
Stevens-Johnson syndrome, the patient may need to use antiviral medication to prevent a recurrence.
When less than 10% of the skin is affected, about 1 to 5% of people with Stevens-Johnson syndrome will
die. With 30% or more skin involvement, the death rate is between 25 to 50%. Large areas of skin can be
lost in days but this process can stop and skin can regrow a few days later.
The risk of death can be estimated with the SCORTEN scale, which assigns one point for each of the
following seven prognostic risk factors: 1) age older than 40 years, 2) heart rate greater than 120 beats
per minute, 3) the presence of associated malignancy, 4) more than 10% of the body surface with skin
detachment or compromise, 5) blood glucose (sugar) levels more than 250 milligrams, 6) BUN (blood urea
nitrogen, which is a measure of kidney health) level more than 27 mg, and 7) sodium bicarbonate level in
the blood greater than 20. The death rate based on the total number of points on this scale is follows: a)
0-1 points, =3.2%, b) 2 points, =12.1%, 3) 3 points, =35.3%, 4) 4 points, =58.3%, and 5) 5 or more
points, =90%.
Regarding the extent of skin reaction, one study found that this was a risk for death in the first 90 days
after the initial reaction but not afterwards. In the same study, age and serious associated medical
problems were risk factors for death between 91 days and 1 year. Of the patients studied, which included
those with TEN, 23% had died at 6 months and 34% had died at one year.
Other factors that lead to a poor prognosis in Stevens-Johnson syndrome is neutropenia lasting more
than five days, hypoalbuminemia (usually less than 2 grams), and persistent azotemia. Neutropenia is a
low number of neutrophils, which is a type of white blood cell that helps protect the body against diseases
and fight infections. Hypoalbuminemia is a low level of albumin in the blood. Albumin is the most abundant
protein in the body. Azotemia is a condition of abnormally high levels of nitrogen-containing compounds in
the blood, usually due to poor kidney filtering. Sepsis and bacteremia (presence of bacteria in the blood)
appear to play an important role in increasing the death rate. Sepsis is a possibly deadly medical
condition characterized by inflammation of the body due to severe infection.
Besides death, other adverse problems from Stevens-Johnson syndrome include blindness (occurs in as
much as 40% of cases), organ damage/failure, and scratching and scarring of the cornea. The latter can
cause inverted eyelashes, sensitivity to light, burning and watery eyes, dry eyes, and excessive blood
vessel growth in the conjunctiva.
If Stevens-Johnson syndrome re-occurs, it is usually more severe and it can be fatal in many cases.
Survival often depends on how much skin damage has occurred and how badly infected the patient has
become.
WHAT IS THE TECHNICALMECHANISM BEHIND SKIN DESTRUCTION IN STEVENS-JOHNSON
SYNDROME?
As noted above, people who have a liver that cannot completely detoxify reactive chemical byproducts of
medications are at increased risk for Stevens-Johnson syndrome. These chemical byproducts may have
a toxic effect or may act as substances that elicit the production of antibodies against the skin. This
makes the skin the target of the antibodies. Cells in local tissue can produces tumor necrosis factor
(TNF)-alpha, which is a type of cell-signaling protein, responsible for widespread inflammation in the body.
When TNF-alpha is produced and the skin becomes a target, T-cells (e.g., CD8+ lymphocytes) are
recruited and increased in number and other types of cells (known as immune effector cells; one of which
is known as a killer effector molecule) are better able to kill cells they target, such as skin cells. It can do
this by releasing certain enzymes (e.g., granzyme B) and a protein (e.g., perforin), which attacks the target
cells. An enzyme is a type of protein that helps produce chemical reactions in the body. The above is one
mechanism for destruction of the top layer of skin. Perforin kills cells by forming tubular structures and
polymers in them. Polymers are large molecules composed of many repeated subunits.
Skin death in the outer layer can also take place when a surface death receptor (also known as cell death
receptors) is activated when they combine with specific types of molecules (e.g., interferon gamma). Both
can be present on the surface of cells on the outermost layer of skin. Surface death receptors (one of
which is known as Fas) are cells that lead to programmed cell death. This is done by triggering activation
of the caspase system, which is a family of enzymes that send out messages that result in the
disorganization of DNA (deoxyribonucleic acid) and leads to cell death. DNA is a chain of many connected
genes. Genes contain coded instructions for how proteins should be constructed and for how certain
bodily characteristics should develop. Researchers have found increased levels of Fas before the skin
detaches and before the onset of damage to mucous membranes.
Once the top layer of skin begins to die and detach from the skin layer beneath it, this triggers the
recruitment of more chemokines, which can continue the inflammation process and result in more
widespread skin death of the outer layer. Chemokines are small signaling proteins secreted by cells.
WHAT ELSE IS STEVENS-JOHNSON SYNDROME KNOWN AS?
Stevens-Johnson syndrome is also known as erythema multiforme major, Stevens-Johnson-Fuchs-
syndrome, syndroma muco-cutaneo-oculare Fuchs, and Fiessinger-Rendu-syndrome, dermatostomatitis
(Stevens Johnson type), ectodermosis erosiva pluriorificialis, erythema multiforme exudativum, erythema
polymorphe (Stevens Johnson type), febrile mucocutaneous Syndrome, Stevens Johnson type, herpes
iris, Stevens-Johnson type, and Johnson-Stevens disease.
WHO ARE SOME FAMOUS PEOPLE WHO HAD STEVENS-JOHNSON SYNDROME?
Some famous people who have had Stevens-Johnson syndrome include a) Manute Bol (a 77 NBA player
who died from complications of the condition), b) Gene Sauers (professional golfer who had to stop
playing from 2006 to 2010 due to the condition), c) Tessa Keller of the MTV show Laguna Beach, d)
Padma Lakshmi (actress, model, and writer; she had the condition at age 14), and e) Ab-Soul (hip hop
artist; he had the condition as a child).
WHAT ARE OTHER NOTABLE CASES OF STEVENS-JOHNSON SYNDROME?
There is the case of 11-year-old Sabrina Brierton Johnson, whose family unsuccessfully tried to sue the
manufacturer (Johnson & Johnson) of Childrens Motrin for one billion dollars after she was blinded by
Stevens-Johnson syndrome after using the medication to treat a fever. The family alleged that Johnson &
Johnson failed to adequately inform people of the risk of developing Stevens-Johnson syndrome from
using the medication. The jury reached their decision in 2008, deciding that the failure to warn was not the
cause of her blindness. The plaintiff was unable to prove that ibuprofen (the active ingredient in Childrens
Motrin) was the cause of Stevens-Johnson syndrome because she also had herpes and chicken pox,
which also could have caused the condition.
There is also the widely reported case of Calvin Locke, who lost 65% of his skin and almost died from
Stevens-Johnson syndrome (or TEN) after using Nurofen, another childrens medication containing
ibuprofen.
WHY IS IT CALLED STEVENS-JOHNSON SYNDROME?
Stevens-Johnson syndrome is named after Drs. Albert Mason Stevens and Frank Chambliss Johnson,
American pediatricians who published a description of the disorder in the American Journal of Diseases
of Children in 1922. The article was entitled A new eruptive fever associated with stomatitis and
ophthalmia; report of two cases in children. The two children were ages 7 and 8 and had extensive
involvement of the mouth and eyes, along with fever. Both cases had been misdiagnosed by their primary
care physicians as hemorrhagic measles. Hemorrhagic refers to bleeding. Measles is a viral illness that
leads to a fever and a characteristic rash. Stevens and Johnson suspected an unknown type of infection
as the cause. They did not consider it to be a case of erythema multiforme because of the lack of
subjective symptoms, the type of skin lesions, the length of the high fever, and the heavy crusting.